Lipid Transport and Blood Lipid Levels
Learning Objectives
Explain why lipoproteins are necessary for triglyceride and cholesterol transport.
Describe the main carriers of cholesterol and triglyceride throughout the body, including how their apolipoproteins affect their endocytosis or catabolism.
Apply your knowledge of cholesterol transport to explain why someone may have changes in HDL and LDL levels.
Understand the etiology of high cholesterol and its potential role in atherosclerosis.
Apply your understanding of cholesterol absorption, synthesis and transport to evaluate the relationships between dietary cholesterol and triglycerides and cardiovascular risk.
Explain the role of lipoprotein lipase in lipid transport, including how it is regulated.
Key Concepts and Vocabulary
Lipoprotein particles including:
Chylomicrons
Very Low Density Lipoproteins (VLDL)
Low Density Lipoproteins (LDL)
High Density Lipoproteins (HDL)
Apolipoproteins, especially ApoB48, ApoB100 and ApoCII
Lipoprotein Lipase and its regulation
Reverse Cholesterol Transport
Trans-Intestinal Cholesterol Export (TICE)
Fatty Acid Transport and Albumin
Triglyceride and Fatty Acid Transport Mechanisms
Transportation of lipids presents some logistical problems. Since they are inherently insoluble, lipids need to be either solubilized prior to transport to other tissues via the blood stream. This is accomplished in two ways. One is the packaging of triglycerides and cholesterol esters into lipoprotein particles, such as the chylomicrons discussed earlier in this unit. The second mechanism is to break triglycerides down to fatty acids, where they can bind to solubilizing proteins called albumin within the blood.
Lipolysis and Fatty Acid Transport
As we described in the unit about lipid oxidation, the majority of our triglyceride stores are in adipose tissue. The release of free fatty acids and glycerol from adipose tissue is a highly regulated process, activated by adrenaline and inhibited by insulin (for more details see Zechner et al. (2012) for a review). The transport of free fatty acids after release from adipose tissue is mediated by albumin, a very abundant protein produced by the liver. Due to their semi-solubility, fatty acids also require transport systems and fatty acid binding proteins (abbreviated as FABP) to move through membranes and through the cytoplasm.
Lipoprotein Particles in the Body
In terms of moving triglycerides and cholesterol esters, we have a variety of lipoprotein particles that play different roles in the body. These are summarized in Table ↑. There are three main transport routes. The first is from the enterocyte to the periphery, mediated by chylomicrons. The second is from the liver to the periphery, mediated by VLDL. The third is from the periphery back to the liver, mediated by HDL and LDL. We will discuss each of these in the next few sections.
| ccc@ Particle | Source | Destination |
|---|---|---|
| Chylomicron | Enterocyte | Adipose, Muscle, Liver |
| VLDL | Liver | Adipose, Muscle |
| IDL | VLDL | Liver or LDL |
| HDL | Endothelial | LDL |
| LDL | IDL/HDL | Liver |
The goal of these lipoprotein particles is to move lipids from the sourceThe source of chylomicrons transporting dietary lipids is the enterocyte, or the source of VLD is the liver; the function of lipoproteins is to transport the lipids from both these sources, enterocyte and liver to peripheral tissues which might be better equipped to utilize or store these lipids. As summarized in Table ↑, these particles are characterized by distinct lipoproteins.
| lccccc@ % Changed to 6 columns Particle | ApoA | ApoB | ApoC | ApoE |
|---|---|---|---|---|
| Chylomicron | AV | B48 | CII/CIII | E |
| VLDL | AV | B100 | CI/CII | E |
| IDL | B100 | E | ||
| HDL | AI/AII | E | ||
| LDL | B100 |
The Role of Chylomicrons and VLDL
Both chylomicrons and VLDL function to move lipids to peripheral tissues, either from the gastrointestinal tract or the liver, respectively. These particles transport primarily neutral lipids rather than free fatty acids. Their assembly is dependent on the production of the apolipoproteins and the presence of phospholipids, especially phosphatidylcholine that is important for their synthesis. If choline levels are limited, either due to less active variants in PEMT or reduced choline dietary intake, the liver will be less able to assemble VLDL. This can result in increased hepatic steatosis, potentially leading to metabolic dysfunction associated fatty liver disease.
The Role and Regulation of Lipoprotein Lipase
Both VLDL and chylomicrons are targeted to peripheral tissues. This specificity is mediated by Apolipoprotein CII. This protein acts as an activator of a triglyceride lipase known as Lipoprotein Lipase or LPL. This lipase resides on the lumen of blood vessels, adjacent to muscle and adipose tissues. Once activated by ApoCII binding, LPL breaks down the triglycerides in the particle and releases its free fatty acid content. These free fatty acids enter the cell where they can be stored (if they enter into the adipocytes), or are used as fuel (if they enter into the muscle cells). The levels of LPL are inversely regulated in adipose and muscle tissue. For example, insulin promotes LPL transcription in adipose tissueTo promote lipid storage. This is accomplished by both transcriptional and post-translational mechanisms, reviewed in Kersten (2014). but decreases LPL transcription in muscle (Spooner et al. 1979). The inverse is true during fasting, where the muscle LPL transcription is increased while that of the adipose tissue is reduced thus promoting muscle uptake of fat to be used as energy, while limiting adipose tissue ability to store this fat in times of energy need (fasting).
LPL is inactivated by diets high in saturated fats. This means that when saturated fat levels are increased, LPL activity is reduced. You can imagine how this is no beneficial to the body, as the lipids in the blood cannot be taken up into the muscles or adipose tissue, therefore remaining in the blood and circulation. This is a negative feedback mechanism wherein intracellular lipids can signal to the LPL on the extracellular surface to prevent additional fat uptake (see Figure ↑). The molecular underpinnings of this phenomena have recently been determined and involve the transcriptional activation of a protein called ANGPTL4Unhelpfully, an abbreviation for Angiopoietin-like 4.. ANGPTL4 is induced when excess fatty acid levels in the cell activate the transcription factor PPAR\(\alpha\). ANGPTL4 is then secreted where it binds to and inhibits the activity of LPL (more details about this can be found in the recent review by Dijk and Kersten (2014)). Mutations in either the LPL or ANGPTL4 genes result in either impaired, or enhanced blood lipid clearance, respectively and as a result either lead to an increased or decreased risk of cardiovascular disease (Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators et al. 2016).
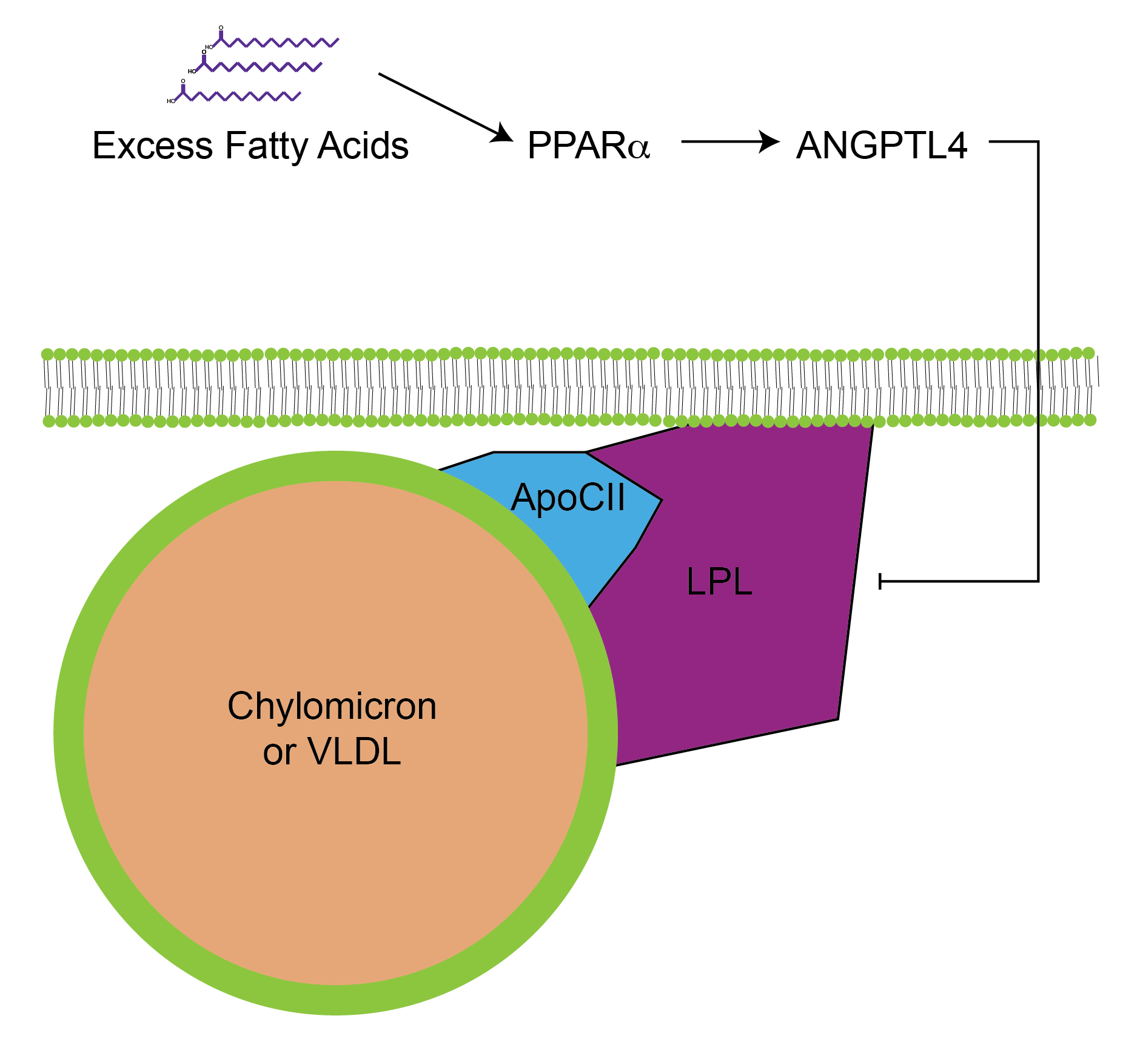
Depleted VLDL are known as IDL, whereas depleted chylomicrons are known as chylomicron remnants. Once these lipoproteins have delivered their triglyceride content into cells, they are either known as chylomicron remnants or intermediateThis indicates that this lipoprotein has intermediate density that falls in between VLDL and LDL’s density density lipoproteins (IDL). Due to the presence of ApoE on their surface, chylomicron remnants and IDL particles can be absorbed by the liver where the apolipoproteins and phospholipids can be reused.
APOE variants are associated with disease risk. ApoE is present on both chylomicrons and the VLDL that then form IDL/LDL. This gene is the major heritable risk factor for late onset Alzheimer’s disease. There are four variants of the APOE gene numbered 1–4These are variants of the same gene, not different genes. Of these isoforms, ApoE2 is thought to be protective of Alzheimer’s disease and associated with lower levels of LDL-cholesterolThis appears to be specific to the APOE gene, as other genetic factors that have more pronounced effects on LDL-cholesterol do not have as strong an association with AD. while ApoE4 is a strong risk factor for late-onset Alzheimer’s disease, and is associated with higher levels of LDL-cholesterol (Poirier et al. 1993; Corder et al. 1993).
Reverse Cholesterol Transport
Cholesterol is primarily disposed of via bile salt generation and excretion, a process that starts in the liver. Therefore cholesterol, which is made throughout the body, is primarily trafficked to the liver, a process known as Reverse Cholesterol Transport. This process is mediated by both HDL and LDL particles. To separate blood lipids between reverse and forward transport processes, sometimes the ratio betwen ApoB and ApoAI is determinedConsider based on the data in Table ↑ what this ratio is actually measuring. As a hint, a high ApoB100/ApoA1 ratio is indicitave of elevated cardiovascular risk.. An alternate cholesterol disposal pathway is through the intestine, a process known as trans-intestinal cholesterol excretion (TICE). In this pathway, cholesterol is transported by either HDL or LDL directly to the intestine for efflux. Estimates vary, but we think that somewhere between 20 and 40% of cholesterol excretion is through TICE, with the remainder being through biliary transport (Temel and Brown 2015).
Synthesis and Role of HDL
High density lipoprotein particles start off as nascent particles containing ApoAI, ApoAII and very little cholesterol. As they pass through the circulation, they bind cholesterol from the plasma membrane of tissues and become enriched with cholesterol. Since most tissues make but cannot dispose of cholesterol, HDL is important for scavenging excess cholesterol from our cells. The HDL particles may be endocytosed in the liver where cholesterol can be disposed, but most often they transfer their cholesterol to LDL particles using an enzyme known as cholesterylester transfer proteinAbbreviated as CETP. This ensures that triglycerides are packaged in the LPL-accessible particles for peripheral transport to be used by peripheral tissues, while excess cholesterol is delivered back to the liver or intestine for excretion. Inhibition of CETP results in an increase in the amount of HDL cholesterol in the blood and thus CEPT inhibition was a heavily invested pharmacological area, but these drugs have shown limited cardiovascular benefits. The current thinking in this area is that high HDL cholesterol is a marker of lowered cardiovascular risk but does not cause lowered cardiovascular risk by itself.
LDL-mediated Transport to the Liver
Low density lipoproteins are generated when IDL derived from VLDL remains in the circulation, or when cholesterol is passed from an HDL to an IDL particle. These particles tend to be cholesterol rich, since the triglycerides have been already taken up due to the actions of LPL at peripheral tissues. These particles would normally be endocytosed by the liver where LDL receptors are found. LDLR levels are under control of the SREBP2. Recall that when intrahepatic cholesterol levels are high, SREBP2 is inactive, and thus LDLR would not be produced. This means that when the liver has sufficient cholesterol, LDL particles remain in the circulationAs a thought exercise, consider what would happen if you had a LDLR mutation, how would that affect cholesterol retrieval? How do you think it would affect cholesterol synthesis? This is the case for individuals with a disease known as familial hypercholesterolemia.. LDL cholesterol levels are correlated with coronary events. If one wants to understand the levels of LDL cholesterol in a person there are two measures to consider. By looking at how much cholesterol is in the LDL fraction (LDL-C, should be less than 100 mg/dL) or how many LDL particles are present in the blood. Since each LDL contains one, and only one ApoB100 particle, and since LDL particles vastly outnumber VLDL particles, then if you determine the concentration of ApoB100 in blood, then that is a measure of LDL particle number. Recent studies have suggested that it is this particle number, more so than the amount of cholesterol in the LDL fraction, that is more predictive of cardiovascular events, though generally both the cholesterol content in LDL and the LDL particle number incresae for most people simultaneously (Cromwell et al. 2007; Mora et al. 2007).
Cholesterol Export to Bile
Within the liver, bile salt synthesis is controlled by the activity of 7-\(\alpha\)-hydroxylaseWe discussed this in the lipid digestion lecture and exported to the gallbladder for release into the digestive system. Separate from the SREBP2-dependent cholesterol regulatory system, the production of bile salts is sensed by the FXR sensing systemFXR is a bile acid sensor expressed in entero-hepatic tissues..
Blood Lipids and Cardiovascular Risk
If lipid transport systems (of VLDL and LDL) have exceeded their ability to store lipids, then VLDL and LDL lipids remain in the blood. Akin to the hyperglycemia associated with impaired glucose disposal, and excessive glucose production, hyperlipidemia is associated with cardiovascular disease due to impaired lipid disposal. Since triglycerides can be metabolized into energy by most tissues, but cholesterol cannot, hypercholesterolemia in particular has been long associated with cardiovascular risk (Keys et al. 1963).
Since cholesterol may exist in several lipoprotein particles, a more prognostic indicator of cardiovascular risk is the amount of cholesterol in HDL particles relative to the amount of cholesterol in LDL particles, with the latter being more pathogenicHDL cholesterol levels may indicate a surplus of cholesterol transport particles, whereas LDL cholesterol likely indicates a surplus of cholesterol that cannot be absorbed by the liver.. Several mechanisms for LDL’s specific association with cardiovascular risk have been proposed, but one possibility is that excess LDL is absorbed in blood vessel walls, promoting both atherosclerosisThe lipid-based coating of arteries, causing arteries to become narrower and reducing their vascular flexibility. and increasing the risk of thrombosisThe release of a blood clot, often by lysis and release of an atherosclerotic plaque. This blood clot could travel to the brain or heart where a stroke or heart attack may occur..
From a dietary standpoint, data from several US-based cohort studies demonstrated that diets lower in saturated fat intake are associated with both lower total and LDL cholesterol, and with reductions in cardiovascular disease (Anderson et al. 1987; Wang et al. 2016). This has recently been challenged by a large-multi country studyThis is known as the PURE study, which evaluated over 135 000 participants in 18 countries. which indicated that increased carbohydrate intake plays a key role, maybe more so than saturated fats with respect to cardiovascular disease (Mente et al. 2017). For this large multi-ethnic study, there is some debate about whether regional dietary differences are fully accounted for, or if this is more reflective of diet-disease risk in a more diverse dataset.
Reflection Questions
A patient carries a loss-of-function mutation in ANGPTL4. Predict the effects on: (a) LPL activity after a high-fat meal, (b) circulating triglyceride levels, and (c) the distribution of dietary fatty acids between adipose and muscle. Then explain why population studies have associated ANGPTL4 loss-of-function variants with reduced cardiovascular disease risk.
Patient A has elevated LDL-C but a normal ApoB100 concentration. Patient B has normal LDL-C but elevated ApoB100. Using your knowledge of VLDL-to-LDL progression and what each measurement represents, evaluate which patient is at higher cardiovascular risk and why, and describe what metabolic or genetic conditions could produce each pattern.
A patient carries the ApoE4 allele and has elevated LDL cholesterol. They ask whether reducing dietary cholesterol is the best strategy to lower their LDL. Apply your knowledge of SREBP2 regulation, LDLR expression, and the relationship between dietary and endogenous cholesterol to advise this patient, and explain what interventions targeting different steps in the system might be more effective.
Lipid Unit Integration Questions
A person eats a meal containing a grilled salmon fillet (rich in \(\omega\)-3 PUFA and protein), roasted vegetables (fiber and fat-soluble vitamins), and olive oil dressing (MUFA). Trace the complete fate of the dietary lipids from digestion through absorption, chylomicron assembly, peripheral delivery, and eventual storage or oxidation. Your answer should address bile salt function, LPL regulation, the differential handling of PUFA vs. MUFA, and how the fiber in the meal might modify cholesterol absorption.
A patient with type 2 diabetes and MASLD presents with hypertriglyceridemia, low HDL, and elevated LDL particle number despite a normal LDL-C. Using your knowledge of the entire lipid unit, explain how insulin resistance simultaneously drives: excess VLDL secretion from the liver (via unrestrained de novo lipogenesis and impaired insulin suppression), impaired LPL activity in muscle, unrestrained lipolysis in adipose, and how excess hepatic Acetyl-CoA from both fatty acid oxidation and de novo synthesis contributes to the overall picture.
An endurance athlete on a very low carbohydrate ketogenic diet completes a four-hour training ride. Trace the metabolic events from adipose lipolysis through fatty acid transport (albumin, LPL, CPTI), \(\beta\)-oxidation, hepatic ketogenesis, and peripheral ketolysis in muscle. Include the role of malonyl-CoA in preventing futile cycling, explain why the brain can shift to using ketone bodies, and calculate (approximately) the energetic cost of using ketone bodies rather than direct glucose oxidation.
A patient with familial hypercholesterolemia (loss-of-function LDLR mutation) is started on a statin plus ezetimibe combination. Using your knowledge of cholesterol synthesis (HMGCR, SREBP2), intestinal absorption (NPC1L1, SREBP2 in enterocytes), bile acid cycling (enterohepatic circulation, fiber), and LDL particle clearance (LDLR), evaluate the mechanism of each drug, predict their combined effect on LDL-C, and explain why combination therapy is more effective than either drug alone.
Four dietary or lifestyle interventions have been proposed to reduce cardiovascular disease risk: (1) reducing dietary cholesterol intake, (2) replacing saturated fatty acids with monounsaturated fatty acids, (3) increasing soluble fiber intake, and (4) improving insulin sensitivity through exercise or weight loss. Using your knowledge of cholesterol absorption (NPC1L1, SREBP2), synthesis (HMGCR), lipoprotein metabolism, and the role of insulin in lipid homeostasis, compare and contrast the mechanistic basis and expected effectiveness of each intervention on LDL-C and cardiovascular risk. Which intervention(s) would you prioritize for a patient with type 2 diabetes and elevated LDL particle number, and why?
Chronic alcohol consumption is associated with hypertriglyceridemia, elevated LDL-C, and increased cardiovascular disease risk. Using your knowledge of alcohol metabolism (ADH vs. CYP2E1 pathways), hepatic NADH generation, effects on TCA cycle activity, de novo lipogenesis, VLDL secretion, and oxidative stress on LDL particles, trace the mechanistic pathway from alcohol consumption to cardiovascular risk. Your answer should distinguish between moderate and heavy drinking, and explain why the shift from ADH to CYP2E1 in heavy drinkers worsens the lipid phenotype.