Regulation of Lipid Catabolism
Learning Objectives
Explain how triglyceride breakdown into glycerol and free fatty acids is controlled in adipocytes by hormonal signals.
Explain how high carbohydrate diets affect fuel utilization, including effects on lipid fuel utilization. Describe at an endocrine level how this is thought to occur.
Determine how much energy, in ATP equivalents, is released during the oxidation of a given fatty acid. Be able to relate the energy content of a fatty acid, in general to its physical properties (length and saturation).
Explain the rate limiting steps of lipid oxidation.
Explain how ketone bodies are converted to ATP in non-hepatic tissues, and what governs this specificity.
Demonstrate an understanding of how how de novo lipogenesis and \(\beta\)-oxidation are reciprocately controlled.
Describe how very long chain fatty acids are oxidized differently from long chain fatty acids.
Explain how odd-numbered fatty acids are catabolized, including the importance of vitamin B12 in this process.
Evaluate the role of transcriptional regulation and long term adaptations to fatty acid oxidative capacity.
Lipolysis Liberates Fatty Acids from Triglyerides
Fatty acids are generally stored as triglycerides, and those triglycerides are primarily stored in adipose tissue. There are two main sources of fatty acids, the dietary fatty acids liberated from chylomicrons by the activity of lipoprotein lipaseThis will be described in more depth in the lipid transport lecture, and fatty acids liberated from adipose tissueA quantitatively small amount of lipids may be stored in liver (too much of this is called hepatic steatosis) and muscle (intramuscular triglycerides) tissues as well. This step, the conversion of triglycerides to fatty acids is known as lipolysis.
In adipocytes the most potent activators of lipolysis are catecholamines such as adrenaline and cortisol. Adrenaline functions to rapidly activate adipocyte triglyceride lipolysis to glycerol and fatty acids via the activation of two enzymes, adipocyte triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL). These two enzymes work primarily on triglycerides and diglycerides, respectivelyThe last step, the conversion of monoacylglycerol to glycerol and a fatty acid, is catalyzed by monoacylglycerol lipase, an enzyme not thought to be regulated by hormones.. Via PKA-dependent signaling, both HSL and ATGL have increased rates of activity resulting in fatty acid release for use as energy in peripheral tissues. Cortisol also increases lipolysis, potentially through several mechanisms, one of which is increasing the levels of ATGL, in a slower more permanent manner.
Carbohydrate Overfeeding and Lipid Utilization
Insulin potently restricts adipocyte lipolysis. The mechanisms of this are still not clear, but at least part of the actions of insulin are to inactivate the lipases HSL and ATGL by reversing the phosphorylation events caused by adrenaline/PKA. States of insulin resistance such as type 2 diabetes means that insulin is less able to keep lipids in the adipose tissue. Interestingly, studies where carbohydrates are given in large quantities as part of overfeeding experiments lead to fat accumulation (Acheson et al. 1988). This was originally thought to be due to activated de novo lipogenesis, but more intricate studies showed that instead, the body was adapting to high levels of carbohydrates by sparing fat (McDevitt et al. 2001) and that the level of lipogenesis was minimal. This lipid sparing effect is the result of increased glucose oxidation and reduced lipolysis and \(\beta\) oxidation. This is a concept we have discussed several times already in this class, when one macromolecule is in relative excess, we tend to alter our metabolic pathways to use this for fuelSome examples we have already discussed include the inhibitory effects of Alanine on Pyruvate Kinase, Citrate on PFK1 and Palmitate on Acetyl-CoA Carboxylase.. Of the multiple potential mechanisms for this lipid sparing effect, one is that carbohydrates induce a strong insulin response, which prevents lipolysis and results in lipids being trapped in adipose tissue. This distinction is key to understanding how high carbohydrate diets relate to obesity.
Fatty Acid Oxidation
Fatty acid oxidation is important in many contexts, including during endurance exercise, and fasting. It is particularly important in the heart where even under resting or elevated carbohydrate conditions, the majority of ATP is derived from fatty acid oxidation (Neely and Morgan 1974). Once fatty acids enter or are made available to the cell, they are quickly converted to "activated" forms known as acyl-CoA moleculesThe acyl refers to a generic fatty acid molecule, so the specific end product could be oleyl-coA (from oleate) or palmitoyl-CoA (from palmitate).. This is also the first step for triglyceride esterification, and consumes two ATP equivalents. The enzymes that catalyze these reactions are known as Acyl-CoA Synthetases. More details about these enzymes can be found in this recent review: (Grevengoed et al. 2014).
\[\begin{equation} \label{eq:acs} Fatty Acid + CoA + ATP \rightarrow AcylCoA + AMP + PPi \end{equation}\]
While there are several Acyl-CoA Synthetases, one in particular, ACSL1Abbreviated from Acyl-CoA Synthetase Long Chain Family Member 1, indicating a preference for long chain fatty acids seems particularly important for fatty acid oxidation, as its genetic ablation in mice almost completely prevents fatty acid oxidation (Ellis et al. 2011). Interestingly, ACSL1 is present on the surface of the mitochondria in a physical complex with CPTI, the most important regulatory protein in fatty acid oxidation (Lee et al. 2011).
Mitochondria are key for fatty acid oxidation. Like most amino acid catabolism, fatty acid conversion to energy requires mitochondria. However, unlike both pyruvate and most amino acids, the transport into the mitochondria is the rate-limiting step for fatty acids. This transportWhich is more difficult than passive diffusion, due to the semi-insoluble nature of fatty acids is controlled by the carnitine shuttle system.
The Role of CPTI in Lipid Oxidation
The carnitine shuttle system is a mechanism to get long chain fatty acidsShort and medium-chain fatty acids can transport through the membranes freely, and therefore are not subject to this system. This also means that they are not subject to regulation via malonyl-CoA-dependent inhibition. through both the outer and inner mitochondrial membranes. It comprises of two reactions and a transporter. The reactions are overall energetically neutral, with the end products identical to the initial reactants. The two reactions are catalyzed by Carnitine Palmitoyltransferase I (CPTI):
\[\begin{equation} AcylCoA + Carnitine \rightarrow Acyl-Carnitine + CoA \end{equation}\]
and Carnitine Palmitoyltransferase II (CPTII):
\[\begin{equation} Acyl-Carnitine + CoA \rightarrow AcylCoA + Carnitine \end{equation}\]
There is a transporter on the inner mitochondrial membrane called Carnitine Acyltransferase (CAT). This transporter only works with the carnitine conjugated fatty acids, so this transport system requires carnitine for its activity. As we will discuss in the non-protein compounds made from amino acids unit, carnitine can be made endogenously from the essential amino acid lysine. Carnitine can also be obtained in the diet, with particularly high levels in red meat.
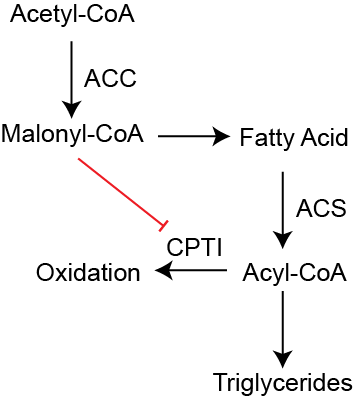
Malonyl-CoA inhibits CPTI. This is the most important regulatory step in fatty acid oxidation. If Acetyl-CoA Carboxylase (ACC) is activated due to an excess of Acetyl-CoA, Citrate or insulin signalingRefer back to the lipid synthesis notes/lecture for more information, then there is a buildup of its product Malonyl-CoA in the cytoplasm. This product potently inhibits CPTI (McGarry et al. 1977). The result of this is to prevent fatty acid import into the mitochondria and therefore fatty acid oxidation. The activated fatty acids are then shuttled towards triglyceride synthesis rather than oxidation. This regulatory process is illustrated in Figure ↑. The flip side of this inhibition is that when AMPK or PKAVia activation by adrenaline or glucagon for example. are active, ACC is inhibited, Malonyl-CoA is reduced and CPTI is active. In this way, energy demand or adrenaline can promote fatty acid oxidation.
Lipid Oxidation in the Mitochondria
Once the Acyl-CoA is inside the mitochondria, oxidation occurs through a series of enzymatic reactions. These reactions proceed in a serial manner to extract one Acetyl-CoA at a time, removing two carbons each cycle. The cycles consist of the following:
- Desaturation
-
This generates a double bond at the \(\Delta^2\) position of the fatty acidThe Co-A group is attached at the carboxyl group, or the \(\Delta^1\) position.. This step generates one FADH\(_2\) molecule.
- Isomerization
-
This transfers the electrons from this new double bond to a new ketone group on the \(\Delta^3\) carbon. This step generates one NADH molecule.
- Release
-
The first two carbons are now released as Acetyl-CoA by an enzyme called a thiolase.
After one round, we are left with a fatty acid that is two carbons shorter, and have generated one Acetyl-CoA, one NADH and one FADH\(_2\) that can be used as fuel. For a saturated fatty acid such as palmitate (C16:0) this means that this cycle occurs seven timesNot eight, because the last reaction ends with two Acetyl-CoA molecules..
Alternative Fatty Acid Catabolism
Long chain, unsaturated fatty acids are catabolized in a very similar manner with one difference. The first desaturation step introduces a double bond, but for unsaturated fatty acids the double bond is already present. That means that during progressive oxidation, if a double bond is already present at the \(\Delta^2\) of the now shortened fatty acid the first step is skipped, and FADH\(_2\) is not generated.
Odd-numbered fatty acids after the last thiolase step, you end up with an Acetyl-CoA and a three carbon Propionyl-CoASeveral amino acids, including Isoleucine, Valine, Threonine and Methionine also produce Propionyl-CoA. Propionyl-CoA is then converted to Succinyl-CoA by a three step reaction that in net consumes one ATP and requires Vitamin B12. Succinyl-CoA is a component of the TCA cycle, and can be further oxidized generating one GTP and one NADH.
Very long chain fatty acids are first oxidized in the peroxisome. The Acyl-CoA Synthetase with activity towards very long chain fatty acidsThose with more than 22 carbons. resides on the peroxisomes. Peroxisomes are membrane enclosed organelles that contain similar enzymes to those that perform \(\beta\)-oxidation in the mitochondria. The major difference is that while these lipids are catabolized, there is no electron transport chain in peroxisomes, so no ATP is produced. Instead electrons are transferred to peroxide, which is in turn converted to water and oxygen by the enzyme Catalase. This process terminates with short and medium chain fatty acids that are released as acyl-carnitines where they travel to mitochondria for final catabolism and some energy production.
Determining the Energy Content of a Fatty Acid
Based on the series of reactions above, or each two-carbon unit released, one Acetyl-CoA, one NADH and one FADH\(_2\) are released. Added together this provides 14 ATP equivalents per two carbon units. Therefore to calculate the ATP in a saturated fatty acid like palmitate (C16:0) we could do the following:
Consume 2 ATP to activate the fatty acid (see the ACS reaction ↑).
Take the fatty acid length, and subtract 2 then divide by 2. This is the number of fatty acid oxidation cycles. For palmitate that is \(\frac{16-2}{2}=7\). For each cycle we generate 14 ATP units, for palmitate there is \(14 x 7 = 98\) ATP equivalents generated.
The remaining Acetyl-CoA from the last two carbons left adds another 10 ATP equivalents
The total ATP is then \(98+10-2\) or 106 ATP equivalents.
For unsaturated fatty acids, you do not need to perform the desaturation step, so for each double bond there is one less FADH\(_2\) generated than normal. If we had a fatty acid that was 16:1 it would generate a total of 104.5 ATP equivalents.
| cl@ Lipid | Net ATP |
|---|---|
| Butyrate (C4:0) | 22 |
| Laurate (C12:0) | 78 |
| Palmitate (C16:0) | 106 |
| Oleate (C18:0) | 120 |
| Linoleate (C18:2) | 117 |
Generally this procedure explains two bioenergetic properties of fatty acids. Longer chain fatty acids have more energy than shorter chain fatty acids, and saturated fatty acids have more energy than desaturated fatty acids. I have used the method above to calculate the ATP equivalents of several fatty acids in Table ↑ to illustrate this point. Don’t try to memorize these, but rather spend some time practicing how to calculate the ATP-equivalentsFor even more of a challenge, try to calculate the net energy production from an odd-chain fatty acid.. After you have thought about how to calculate this, take a step back. This is a lot of energy storage. A triglyceride with three C16:0 molecules is catabolized to 318 ATP equivalents. This is close to ten times the energy storage of a fully oxidized glucose molecule (32 ATP).
Ketolysis
Ketone bodies are produced when there is excessive acetyl-CoA, but insufficent TCA cycle intermediates in the liver. These ketone bodiesPrimarily \(\beta\)-hydroxybutyrate then acetoacetate; acetone is a terminal end product and cannot be used for energy. can be converted back to Acetyl-CoA in non-hepatic tissues. This specificity is due to the absence of an enzyme called 3-oxoacid CoA-transferase 1OXCT1, sometimes called succinyl-CoA-3-oxaloacid CoA transferase (or SCOT). in the liver thus not allowing the liver to utilize ketone bodies while allowing other tissues to utilize ketone bodies for energy. This enzyme catalyzes this reaction starting with Acetoacetate\(\beta\)-hydroxybutyrate can be interconverted to and from acetoactetate using up one NADH molecule for each acetoacetate generating, and using an NADH for the reverse reaction.:
\[\begin{equation} \label{eq:oxct1} Acetoacetate + Succinyl-CoA \rightarrow Succinate + Acetoacetyl-CoA \end{equation}\]
As a result this generates a single Acetyl-CoA molecule after a thiolase reaction. This reaction is not cataplerotic because both Succinate and Succinyl-CoA are part of the TCA cycle. However bypassing the Succinyl-CoA Synthetase step means that one GTP is not generated during normal TCA cycle progress. Overall, an Acetyl-CoA that is released from the liver as \(\beta\)-hydroxybutyrate costs the liver 3.5 ATP molecules 1 ATP for the ATP-Citrate Lyase step, 2.5 ATP for the NADH used to generate \(\beta\)-hydroxybutyrate. In the recipient tissue, that \(\beta\)-hydroxybutyrate generates 6.5 ATP in netGenerate 10 ATP from the TCA cycle oxidation of Acetyl-CoA, but using 1 NADH converting \(\beta\)-hydroxybutyrate to acetoacetate; and 1 GTP skipping the Succinyl-CoA Synthetase step. So 10 - 3.5 =6.5. Overall, ketone usage means that the original Acetyl-CoA in the liver now only generates 3 ATP equivalents rather than the 10 ATP if it remained in the liver (6.5 ATP generated - 3.5 ATP used in liver to generate the \(\beta\)-hydroxybutyrate = 3 ATP in total). Table ↑ summarizes the energy costs of the three major transport processes we have discussed this year. The inefficiency of these transport processes comes at a cost to performance, but can cause negative energy balance for weight loss. In terms of low-carbohydrate, high fat diets this may explain the slight increase in energy expenditure (100-200kcal/day) observed in relation to low fat, high carbohydrate diets (Hall et al. 2016; Ebbeling et al. 2018).
| ll@ Process | Net ATP Loss |
|---|---|
| Ketone Body Transport | -7 ATP |
| Cori Cycle (Lactate) | -6 ATP |
| Cahill Cycle (Alanine) | -15 ATP |
Regulation of Fatty Acid Oxidation
Short-term Regulation
As we described above, short term regulation is accomplished primarily through the ACC/Malonyl-CoA/CPTI system described in Figure ↑. Two other key regulatory systems to consider in the short term are the flux of fatty acids, either via the regulation of lipolysis, or the regulation of Lipoprotein Lipase activityDiscussed in the lipid transport lecture.. Another regulatory mechanism that might not seem as apparent, is that when there is sufficient non-lipid fuel sources (for example an excess of carbohydrates or amino acids), there will be more Malonyl-CoA and therefore less lipid oxidation. Finally, recall that the products of fatty acid oxidation all require TCA cycle and electron transport chain activity. Since the most important driver of the electron transport chain is energy demand, the final oxidation of all that Acetyl-CoA, NADH and FADH\(_2\) will only occur if there is some energy demand. Alternately, if there is insufficient TCA cycle intermediatesFor example due to cataplerosis in a liver undergoing substantial gluconeoegenesis. the NADH and FADH\(_2\) generated from the partial oxidation of fatty acids might be used for fuel, but the Acetyl-CoA generated from the partial oxidation of fatty acids will undergo ketogenesis for transport to other tissues.
Transcriptional Adaptations for Fatty Acid Oxidation
In terms of athletic performance, increasing the ability to oxidize fatty acids is important for endurance athletes. Highly trained athletes tend to have more mitochondria, and higher expression of enzymes such as Lipoprotein Lipase, CPT1, Acetyl-CoA Synthetase and enzymes of the TCA cycle. One major regulator of these adaptations are transcription factors called PPAR\(\delta\) and PPAR\(\alpha\). In muscle tissues PPAR\(\alpha\) and PPAR\(\delta\) activate the transcription of several lipid transport and oxidation genes. In liver tissues, a similar transcription factor, PPAR\(\alpha\) upregulates many of the same genesActivation of PPAR\(\alpha\) is a promising target to alleviate hepatic steatosis and metabolic dysfunction-associated steatotic liver disease (MASLD), though they have not yet reached the clinic. along with genes involved in ketogenesis (Kersten et al. 2000; Badman et al. 2007). Both of these transcription factors are induced by elevated intracellular fatty acid levels (Keller et al. 1993). The specific natural ligands for the PPAR transcription factors have been difficult to unambiguously identify, but PUFA’s seem to play a key role in activating these transcription factors (Forman et al. 1997). This is potentially one mechanism by which PUFA’s alleviate lipid accumulation.
Interestingly, similar adaptations occur in the context of obesity, likely for the same reasons. The major difference, is that in the absence of energy demand, lipids are only partially oxidized or end up stored as lipids in cells. The buildup of stored, partially oxidized fatty acids is thought to play a role in the pathogenesis of insulin resistance, though the mechansims are not clearly defined.
Reflection Questions
Calculate the net ATP yield from complete oxidation of one molecule of oleate (C18:1\(\Delta^9\)) in muscle, accounting for the activation cost and the reduced FADH\(_2\) generation due to the double bond. Then compare this to the net ATP a recipient muscle cell would obtain if the liver had instead exported that same Acetyl-CoA as \(\beta\)-hydroxybutyrate. Use this comparison to evaluate the energetic trade-off of ketone body transport.
A patient with a primary carnitine deficiency cannot maintain adequate intracellular carnitine levels. Analyze how this would differentially affect oxidation of short-chain vs. long-chain fatty acids, predict the consequences during prolonged fasting, and explain why the heart would be particularly vulnerable in this condition.
A sedentary person and an endurance-trained athlete consume identical high-fat meals after an overnight fast. Evaluate how their capacity to oxidize dietary fatty acids differs at three levels: (a) hormonal control of lipolysis in adipose, (b) CPTI activity and malonyl-CoA regulation in muscle, and (c) transcriptional adaptations via PPAR\(\alpha\)/PPAR\(\delta\). Predict the metabolic fate of dietary fatty acids in each person.